
Phylogenetic trees are a powerful tool in evolutionary biology that allow scientists to visualize the relationships between different species. These trees, often referred to as cladograms, are constructed based on shared characteristics and genetic information. By analyzing the similarities and differences between different species, scientists can create a branching diagram that represents the evolutionary history of organisms.
The main goal of constructing a phylogenetic tree is to determine the common ancestors of different species and understand the patterns of evolution. This information can provide valuable insights into how species have evolved over time and how they are related to one another. By studying phylogenetic trees, scientists can also gain a better understanding of the ecological and morphological changes that have occurred throughout evolutionary history.
When constructing a phylogenetic tree, scientists compare the genetic sequences or physical characteristics of different species to identify similarities and differences. These similarities and differences are then used to create a branching diagram, with each branch representing a common ancestor. The length of the branches can also provide information about the time since the common ancestor existed, with longer branches indicating more time has passed.
What Are Phylogenetic Trees?
Phylogenetic trees, also known as evolutionary trees or tree diagrams, are visual representations of the evolutionary relationships among different organisms or groups of organisms. These trees are constructed based on similarities and differences in genetic or morphological traits, allowing scientists to understand the evolutionary history and relatedness of species.
Phylogenetic trees have a hierarchical structure, with branches representing different species or groups of species. The branches diverge from a common ancestor, depicting the evolutionary divergence and branching out of different lineages over time. The tree can also show the relative recency of common ancestors and the time frame in which different groups diverged from a common ancestor. The length of the branches can represent the amount of evolutionary change that has occurred over time.
Phylogenetic trees are essential tools in the field of evolutionary biology and are used to study a wide range of biological phenomena. They provide insights into the evolution and classification of organisms, help in understanding patterns of biodiversity, and can be used to infer ancestral traits and evolutionary events. These trees also aid in identifying the relationships between different species, determining their common ancestry, and predicting their evolutionary trajectories.
Constructing a Phylogenetic Tree
To construct a phylogenetic tree, scientists gather data on the genetic or morphological traits of different organisms. This data is usually obtained through DNA sequencing, analysis of fossils, or observations of physical characteristics. The collected data is then analyzed using various computational methods, including statistical algorithms and computer programs.
The construction of a phylogenetic tree involves a process called tree building or tree reconstruction. This process involves grouping organisms based on their shared characteristics and using statistical methods to determine the most likely evolutionary relationships between them. The resulting phylogenetic tree is a hypothesis of the evolutionary history of the organisms being studied, representing the best current understanding of their evolutionary relationships.
It is important to note that phylogenetic trees are not fixed or absolute representations of evolution. They are subject to revisions and refinements as new data becomes available and as our understanding of evolution advances. However, these trees provide a valuable framework for organizing and interpreting the vast diversity of life on Earth and helping us unravel the mysteries of our evolutionary past.
Understanding the Basics of Phylogenetic Trees

Phylogenetic trees are graphical representations of the evolutionary relationships between different species or groups of organisms. These trees show the branching patterns and shared ancestry among organisms, providing valuable information about the history and diversity of life on Earth.
In order to understand a phylogenetic tree, it is important to be familiar with some of the basic components and features. One key element is the root, which represents the common ancestor of all the organisms included in the tree. The branches and nodes on the tree depict the divergence and speciation events that have occurred over time.
- The branches on a phylogenetic tree can be either horizontal or diagonal, indicating different types of relationships between species. Horizontal branches indicate a common ancestor, while diagonal branches represent ancestral species that have gone extinct.
- Nodes, on the other hand, represent the most recent common ancestor of the groups of organisms that are connected by that node. Nodes often have labels indicating the time or estimated age of the common ancestor.
It is also important to understand that the length of the branches on a phylogenetic tree does not necessarily correlate with the amount of time that has passed since a divergence event. Branch length can be proportional to evolutionary change, but it is not a direct measurement of time.
Overall, phylogenetic trees provide a visual representation of the relationships and evolutionary history of different species. By studying these trees, scientists can gain insights into the patterns and processes that have shaped the diversity of life on Earth.
The Importance of Phylogenetic Trees in Evolutionary Biology
Phylogenetic trees play a crucial role in understanding the evolutionary relationships between different species. By visually representing the evolutionary history and branching patterns of organisms, phylogenetic trees provide valuable insights into the processes of speciation, adaptation, and genetic diversity over time.
One of the key uses of phylogenetic trees is in determining the relative relatedness between different species. By comparing the similarities and differences in their genetic sequences or physical traits, scientists can construct phylogenetic trees that show the common ancestry and evolutionary divergence of species. This information is essential for understanding how new species arise, how they adapt to different environments, and how they are related to other organisms.
Phylogenetic trees also provide a framework for studying the patterns of evolution and the spread of traits within a lineage. By analyzing the branching patterns and the distribution of traits on the tree, scientists can make predictions about the evolutionary processes that led to the observed diversity of species. This information can be used to study the impact of environmental changes on the evolution of certain traits, the role of natural selection in shaping the genetic makeup of populations, and the patterns of speciation and extinction over time.
The construction and analysis of phylogenetic trees also have practical applications in fields such as medicine and conservation biology. By understanding the evolutionary relationships between different organisms, scientists can gain insights into the spread of infectious diseases, the development of drug resistance, and the identification of potential disease vectors. Phylogenetic trees can also help guide conservation efforts by identifying the most genetically diverse and evolutionarily distinct populations that need to be protected to maintain overall biodiversity.
In conclusion, phylogenetic trees are a powerful tool in evolutionary biology. They provide a visual representation of the evolutionary history and relationships between different species, allowing scientists to understand the processes of evolution, adaptation, and genetic diversity. Moreover, phylogenetic trees have practical applications in diverse fields, from medicine to conservation biology, helping us better understand and protect the natural world.
How to Interpret Phylogenetic Trees
Phylogenetic trees are diagrams that show the evolutionary relationships between different species or groups of organisms. These trees are constructed based on similarities and differences in genetic or physical characteristics among organisms. Interpreting phylogenetic trees can provide valuable insights into the evolutionary history and the relatedness of different species.
1. Understanding the branches: The branches of a phylogenetic tree represent the evolutionary lineage of a species. Each branch represents a common ancestor, and the length of the branch indicates the amount of time that has passed since the species diverged from its common ancestor. Longer branches indicate more time elapsed, while shorter branches indicate less time has passed.
2. Interpreting the nodes: The nodes of a phylogenetic tree represent the points at which species or groups of organisms diverged from a common ancestor. The nodes can be thought of as a hypothetical ancestral species from which multiple descendant species evolved. Each node represents a new branch in the tree and represents a new lineage.
3. Understanding the tips: The tips or terminal nodes of a phylogenetic tree represent the extant or living species. These are the species that have not yet diverged into separate lineages. The tips can be labeled with the names of the species or with other identifiers.
4. Interpreting the distances between branches: The distances between branches in a phylogenetic tree can provide information about the amount of genetic or physical change that has occurred between species. Closer branches indicate more similarity, while farther branches indicate more divergence.
5. Using the keys or legends: Phylogenetic trees often come with keys or legends that provide additional information about the tree. These keys may include color-coded branches or symbols to represent specific traits or characteristics of the organisms.
- Conclusion
Interpreting phylogenetic trees requires a combination of understanding the layout of the tree, the relationships between branches and nodes, and the additional information provided in the keys or legends. By analyzing the tree, scientists can determine the evolutionary history and relatedness between different species, providing valuable insights into the diversity and complexity of life on Earth.
Key Components of Phylogenetic Trees
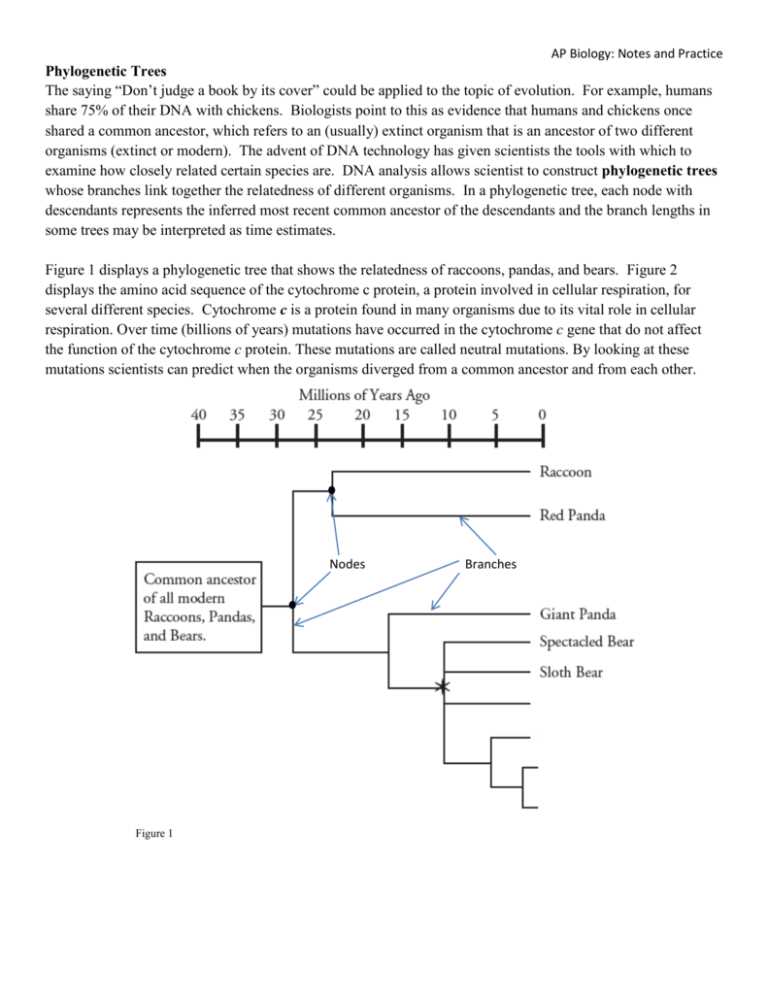
Phylogenetic trees are graphical representations that illustrate the evolutionary relationships among different species or groups of organisms. These trees are constructed based on the analysis of genetic data, such as DNA or protein sequences, and they provide valuable insight into the evolutionary history and relatedness of organisms.
In order to understand phylogenetic trees, it is important to familiarize oneself with some key components. These include:
- Branches: The lines that connect different nodes on a phylogenetic tree are called branches. Each branch represents a different species or group of organisms.
- Nodes: Nodes, also known as branching points, represent the common ancestors shared by different species or groups of organisms. These nodes are typically indicated by a point where multiple branches meet.
- Root: The root of a phylogenetic tree represents the common ancestor of all the organisms or species included in the tree. It is usually depicted at the base of the tree.
- Taxa: Taxa, or terminal nodes, represent the individual species or groups of organisms being compared in the phylogenetic tree. Each taxa is located at the tip of a branch.
- Branch lengths: The lengths of branches in a phylogenetic tree can indicate the amount of genetic change or time that has elapsed since divergence from a common ancestor. Longer branches typically represent greater genetic divergence.
- Characteristics or traits: Phylogenetic trees can also include information about specific characteristics or traits that are shared or unique among different taxa. This information can be used to infer evolutionary relationships and identify common ancestry.
Understanding these key components is essential for interpreting and analyzing phylogenetic trees. They provide a visual representation of evolutionary history and allow researchers to make hypotheses about the relationships between different species or groups of organisms based on their genetic similarities and differences.
Methods for Constructing Phylogenetic Trees
Phylogenetic trees are essential tools in evolutionary biology for understanding the relationships between different species. Constructing these trees involves analyzing various types of data, including genetic sequences and morphological characteristics. Several methods have been developed to create phylogenetic trees, each with its own advantages and limitations.
1. Maximum Likelihood
The maximum likelihood method is widely used in phylogenetics. It compares different models of evolution to find the tree that best fits the observed data. This method takes into account the probability of each possible tree, given the data, and selects the tree with the highest likelihood. Maximum likelihood can handle complex models, including models that account for different rates of evolution among branches.
2. Maximum Parsimony
The maximum parsimony method aims to find the tree that requires the fewest evolutionary changes to explain the observed data. It assumes that the simplest explanation is the most likely. This method constructs a tree by minimizing the total number of evolutionary changes, such as mutations or substitutions, needed to explain the observed data. Maximum parsimony is computationally efficient, making it suitable for large-scale analyses, but it does not consider the rate of evolution on different branches.
3. Bayesian Inference
Bayesian inference is a probabilistic approach to constructing phylogenetic trees. It uses Bayes’ theorem to calculate the probability of different trees given the observed data. This method incorporates prior knowledge or assumptions about the underlying evolutionary process, as well as the observed data, to estimate the posterior probability of each tree. Bayesian inference can handle complex models and has the advantage of providing a measure of uncertainty for the estimated trees.
4. Neighbor-Joining
The neighbor-joining method is a distance-based method for constructing phylogenetic trees. It calculates pairwise distances between taxa based on genetic or morphological data and constructs a tree by iteratively joining the nearest neighbors. This method is computationally efficient and can handle large datasets, but it assumes a constant rate of evolution and may not accurately capture long-branch attraction or other complex evolutionary processes.
In conclusion, constructing phylogenetic trees requires careful analysis of data and consideration of various methods. The choice of method depends on the nature of the data, the complexity of the evolutionary process, and the researcher’s assumptions and objectives. Ultimately, these methods provide valuable insights into the evolutionary history and relationships of different species, helping us understand the diversity of life on Earth.