
Completing a worksheet on sickle cell anemia can be a challenging task, but having the answer key can make it much easier. This article will provide an in-depth explanation of the answer key for a sickle cell anemia worksheet. By understanding the key concepts and information, students will be able to grasp the complexities of this genetic disorder.
The answer key for the sickle cell anemia worksheet contains crucial information about the causes, symptoms, and treatment of this condition. It delves into the genetic inheritance pattern of sickle cell anemia and explains how certain mutations in the hemoglobin gene can lead to the production of abnormal red blood cells. The key will also highlight the prevalence of sickle cell anemia in certain populations, particularly those with a higher prevalence of the sickle cell gene.
Furthermore, the answer key elaborates on the symptoms and complications associated with sickle cell anemia. It will discuss how the abnormal shape of the red blood cells can cause blockages in blood vessels, leading to episodes of pain and potentially more severe complications. The key will also cover the importance of proper management and treatment options for individuals with sickle cell anemia, including the use of medications, blood transfusions, and, in severe cases, bone marrow transplantation.
Sickle Cell Anemia Worksheet Answer Key
Below is the answer key for the Sickle Cell Anemia worksheet:
Question 1:
What is sickle cell anemia?
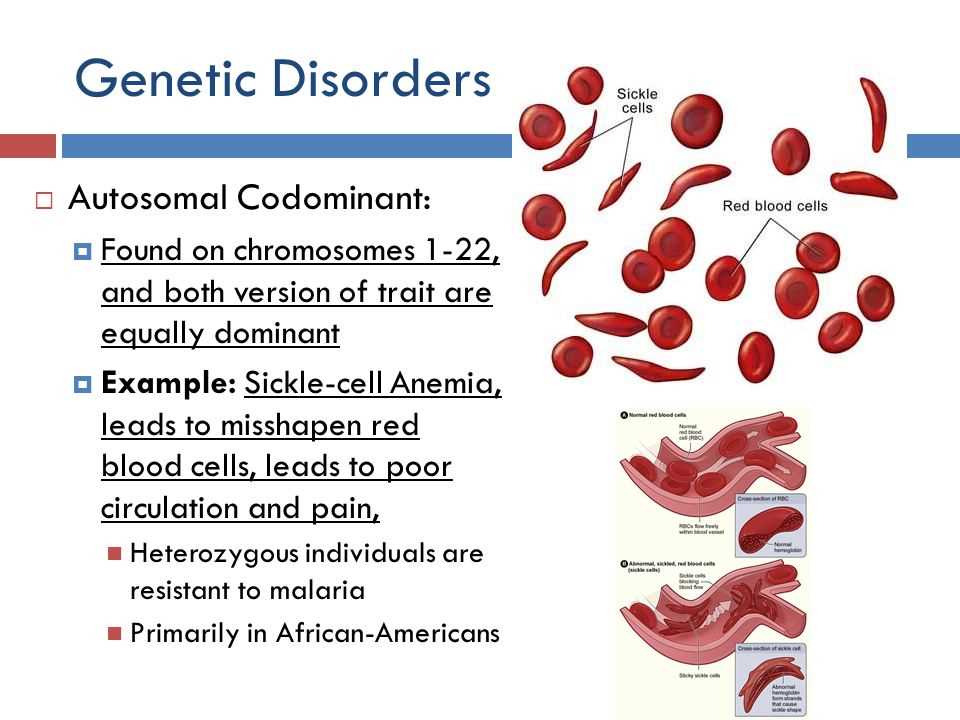
Sickle cell anemia is a genetic blood disorder characterized by the production of abnormal hemoglobin, which causes red blood cells to become rigid and form a sickle shape. This can lead to various complications including pain, anemia, and organ damage.
Question 2:
How is sickle cell anemia inherited?
Sickle cell anemia is inherited in an autosomal recessive manner. This means that both parents must carry a gene mutation for sickle cell in order for their child to inherit the disorder. If both parents are carriers, there is a 25% chance with each pregnancy that the child will have sickle cell anemia.
Question 3:
What are the symptoms of sickle cell anemia?
The symptoms of sickle cell anemia can vary, but common symptoms include fatigue, pain (called sickle cell crisis), shortness of breath, pale skin, and delayed growth in children. Complications can also include infections, stroke, and damage to organs such as the spleen, liver, and kidneys.
Question 4:
How is sickle cell anemia diagnosed?
Sickle cell anemia can be diagnosed through various tests, including a complete blood count (CBC) to check for anemia and abnormal red blood cells, hemoglobin electrophoresis to identify the abnormal hemoglobin, and genetic testing to confirm the presence of the gene mutation.
Question 5:
What are the treatment options for sickle cell anemia?
Treatment for sickle cell anemia focuses on managing symptoms and preventing complications. This may include pain management during sickle cell crisis, blood transfusions to increase oxygen levels in the blood, medication to prevent infections, and hydroxyurea, which can help reduce the frequency of sickle cell crises.
Overall, sickle cell anemia is a complex genetic disorder that requires ongoing management and care to prevent complications and improve quality of life for those affected.
What is Sickle Cell Anemia?
Sickle Cell Anemia is a genetic blood disorder that affects the shape and function of red blood cells. It is caused by a mutation in the gene that produces hemoglobin, the protein responsible for carrying oxygen in the blood. This mutation causes the red blood cells to become stiff and sticky, taking on a crescent or “sickle” shape.
When the red blood cells become sickle-shaped, they are unable to flow smoothly through the blood vessels, leading to blockages and decreased blood flow. This can result in various complications such as severe pain, organ damage, and an increased risk of infections. Sickle Cell Anemia is most commonly found in people of African, Mediterranean, Middle Eastern, and Indian descent.
The symptoms of Sickle Cell Anemia can vary from person to person, but may include fatigue, shortness of breath, jaundice, delayed growth, and frequent infections. The severity of the disease also varies, with some individuals experiencing mild symptoms while others may have frequent pain crises and complications requiring medical intervention.
There is currently no cure for Sickle Cell Anemia, but treatment options are available to manage the symptoms and complications. This may include pain medication, blood transfusions, and bone marrow transplants. Early diagnosis and regular medical care play a crucial role in managing the condition and improving the quality of life for individuals with Sickle Cell Anemia.
Causes of Sickle Cell Anemia
Sickle cell anemia is a genetic disorder that is caused by a mutation in the hemoglobin gene. This mutation leads to the production of abnormal hemoglobin, known as hemoglobin S. The substitution of one amino acid in the beta chain of hemoglobin causes the red blood cells to become sickle-shaped, instead of their normal disc-like shape. These sickle-shaped cells are less flexible and can lead to various complications.
The main cause of sickle cell anemia is inheriting the mutated gene from both parents. This means that both parents must be carriers of the sickle cell trait, which is when a person has one copy of the mutated gene and one copy of the normal gene. When two carriers have a child, there is a 25% chance that the child will inherit two copies of the mutated gene, resulting in sickle cell anemia.
Sickle cell anemia is more common in certain populations, particularly those of African, Mediterranean, Middle Eastern, and Indian descent. This is because the sickle cell trait provides some resistance to malaria, which is prevalent in these regions. As a result, the gene for sickle cell anemia has been selected for over time, leading to a higher prevalence in these populations.
Other factors that can trigger sickle cell crises in individuals with sickle cell anemia include infections, dehydration, extreme temperatures, strenuous exercise, and high altitudes. These factors can cause the sickle-shaped red blood cells to clump together, leading to blockages in blood vessels and reduced oxygen supply to tissues, resulting in pain and organ damage.
Symptoms of Sickle Cell Anemia
Sickle cell anemia is a genetic blood disorder that causes red blood cells to become misshapen and sticky. This can lead to a range of symptoms that vary in severity.
One of the main symptoms of sickle cell anemia is pain. Individuals with the condition often experience episodes of severe pain, known as sickle cell crises. These episodes can last for several days and can occur anywhere in the body. The pain is caused by the blockage of blood vessels by the misshapen red blood cells.
Another common symptom is fatigue. Sickle cell anemia causes the red blood cells to have a shortened lifespan, leading to a shortage of healthy red blood cells. This can result in anemia, which is characterized by fatigue, weakness, and shortness of breath.
In addition to pain and fatigue, individuals with sickle cell anemia may also experience frequent infections. The misshapen red blood cells can impair the immune system’s ability to fight off infections, making individuals with the condition more susceptible to bacterial and viral infections.
Other symptoms of sickle cell anemia can include jaundice, which is yellowing of the skin and eyes due to the breakdown of red blood cells, delayed growth and development in children, and vision problems.
It is important for individuals with sickle cell anemia to seek regular medical care and to manage their symptoms through pain management strategies, blood transfusions, and medications. By properly managing the condition, individuals with sickle cell anemia can live full and productive lives.
Treatment Options for Sickle Cell Anemia
Sickle cell anemia is a genetic blood disorder that affects the red blood cells and can cause severe pain, organ damage, and complications. While there is currently no cure for sickle cell anemia, there are several treatment options available that can help manage the symptoms and improve the quality of life for individuals with the condition.
One of the main treatment approaches for sickle cell anemia is pain management. Painful crises, also known as sickle cell pain episodes, can occur when the sickle-shaped red blood cells block the flow of blood and oxygen to the tissues. Treatment for pain may involve the use of over-the-counter or prescription pain medications, such as nonsteroidal anti-inflammatory drugs (NSAIDs) or opioids. In some cases, opioids may be used for long-term pain management.
In addition to pain management, individuals with sickle cell anemia may benefit from blood transfusions. Blood transfusions can help increase the number of healthy red blood cells in the body and improve circulation. This can reduce the risk of complications and improve overall health. However, frequent blood transfusions can increase the risk of iron overload, so individuals may also receive chelation therapy to remove excess iron from the body.
Another treatment option for sickle cell anemia is hydroxyurea. Hydroxyurea is a medication that can stimulate the production of fetal hemoglobin, which is a type of hemoglobin that can prevent red blood cells from sickling. This medication can reduce the frequency of painful crises and the need for blood transfusions in some individuals with sickle cell anemia. However, it is important to note that hydroxyurea is not suitable for everyone and may have side effects.
Other treatment options for sickle cell anemia may include bone marrow transplantation and gene therapy. These are more invasive procedures and are generally reserved for individuals with severe symptoms or complications. Bone marrow transplantation involves replacing the faulty bone marrow with healthy marrow from a donor, while gene therapy aims to correct the genetic mutation responsible for sickle cell anemia. These treatments can potentially provide a cure for the condition, but they are still considered experimental and may not be widely available.
In conclusion, while there is no cure for sickle cell anemia, there are various treatment options available to manage symptoms and improve the quality of life for individuals with the condition. These treatment options include pain management, blood transfusions, hydroxyurea, and more invasive procedures like bone marrow transplantation and gene therapy. It is important for individuals with sickle cell anemia to work closely with their healthcare providers to determine the most appropriate treatment plan for their specific needs.
Complications of Sickle Cell Anemia

Sickle cell anemia is a genetic disorder characterized by the presence of abnormal hemoglobin molecules in red blood cells. The abnormal shape of the red blood cells can lead to various complications in different parts of the body.
One of the main complications of sickle cell anemia is vaso-occlusive crisis, where the abnormal red blood cells block the blood vessels, causing severe pain and organ damage. This can occur in any part of the body, but most commonly affects the bones, chest, abdomen, and joints. Vaso-occlusive crisis often requires hospitalization and management of pain with medications.
Pulmonary complications
In addition to vaso-occlusive crisis, sickle cell anemia can lead to several pulmonary complications. Acute chest syndrome is a condition characterized by chest pain, cough, fever, and difficulty breathing, and is often caused by infection or pulmonary embolism. This condition can be life-threatening and requires immediate medical intervention.
Pulmonary hypertension is another complication of sickle cell anemia characterized by increased blood pressure in the lungs. This can lead to shortness of breath, fatigue, and eventually heart failure if left untreated. Regular monitoring and proper management of pulmonary hypertension is essential.
Preventing Sickle Cell Anemia
Sickle cell anemia is a genetic disorder that affects the red blood cells and results in abnormal hemoglobin production in the body. It is important to take preventive measures to reduce the risk of sickle cell anemia, especially in individuals who have a family history of the condition or belong to populations with a high prevalence of the disease.
Genetic counseling: One of the key steps in preventing sickle cell anemia is genetic counseling. This involves a trained professional who can assess an individual’s risk of passing on the sickle cell gene to their children. Genetic counseling helps individuals understand the inheritance pattern of the disease and make informed decisions regarding family planning.
Prenatal testing: For couples who are at a high risk of having children with sickle cell anemia, prenatal testing can be an important step in prevention. This involves screening the fetus during pregnancy to determine if they carry the sickle cell gene. Knowing the status of the fetus allows parents to make informed decisions about their pregnancy and potentially take further preventive measures.
Newborn screening: Early detection of sickle cell anemia is crucial for effective management and treatment. Many countries have implemented newborn screening programs that aim to identify infants with the disease shortly after birth. Identifying sickle cell anemia in newborns enables healthcare providers to start early interventions and provide appropriate care to minimize complications.
Vaccinations and preventive measures: Individuals with sickle cell anemia are more susceptible to infections due to their weakened immune system. It is important for individuals with the condition to take necessary vaccinations, such as the pneumococcal vaccine and the annual flu shot, to prevent serious infections. Additionally, individuals with sickle cell anemia should avoid triggers that can lead to a sickle cell crisis, such as extreme cold or dehydration.
Education and awareness: Raising awareness about sickle cell anemia is crucial in preventing the disease. Education programs should focus on increasing knowledge about the inheritance pattern of the disease, the importance of genetic counseling, and the importance of early detection and management. By promoting awareness, individuals and communities can take proactive steps towards preventing sickle cell anemia and improving the quality of life for individuals living with the condition.